1. Introduction
ELISA (enzyme-linked immunosorbent assay) is a plate-based assay for the detection and quantification of peptides, proteins, antibodies and hormones. The ELISA technique is based on an antigen immobilised to a solid surface, which is complexed with an antibody that is linked to an enzyme.
The enzyme activity upon incubation with a substrate and production of a measurable product is used for the detection and quantification of the factor of interest. This detection method relies on the highly specific antibody-antigen interaction. ELISAs are typically carried out in 96-well (or 384-well) polystyrene plates, which will passively bind antibodies and proteins. The binding and immobilization of reagents of interest is easy to design and perform.The subsequent separation of specifically bound from unbound factors makes ELISA assays a powerful tool for the analysis of relevant analytes.
2. General ELISA Procedure
Typically, an ELISA begins with a coating step, in which the first layer, consisting of a target antigen or antibody is adsorbed onto a 96-well polystyrene plate, unless you are using a kit with a plate that is pre-coated with antibody. Following, all unbound sites are coated with blocking agent. After a series of washes, the plate is incubated with enzyme-conjugated antibody. Another series of washes removes all unbound antibody.
The addition of a substrate causes the production of a calorimetric signal, which is finally detected. Because the separation of factors of interest from a heterogeneous sample relies on specific surface binding, several washes are repeated in each ELISA step to remove unbound material. During this process, it is crucial that excess liquid is removed in order to prevent the dilution of the solutions added in the next step. To ensure uniformity, specialised plate washers can be utilised.
An ELISA assay can include multiple intervening steps, particularly when measuring protein concentration in heterogeneous samples such as blood. The detection of multiple antibodies for signal amplification at the same time represents the most complex and varying step in the overall process.

3. ELISA Types
LISAs can be performed as direct, indirect, sandwich or competitive assays. The immobilization of the antigen, the key step of the ELISA method, can be achieved by direct adsorption to the assay plate or indirectly via capture antibody that has been attached to the plate. Further, the antigen is then either detected directly with an enzyme-labeled primary antibody or indirectly by an enzyme-labeled secondary antibody.
The antibodies used for detection are commonly coupled with alkaline phosphatase (AP) or horseradish peroxidase (HRP). Dependent on the required assay sensitivity and the signal-detection method (spectrophotometer, fluorometer or luminometer), a variety of substrates are available for performing the ELISA with an HRP or AP conjugate. Hence, when designing an ELISA assay, the different ELISA strategies, specifically for the detection step, have to be differentiated and adjusted to the requirements of the nature of the sample and the biological question.
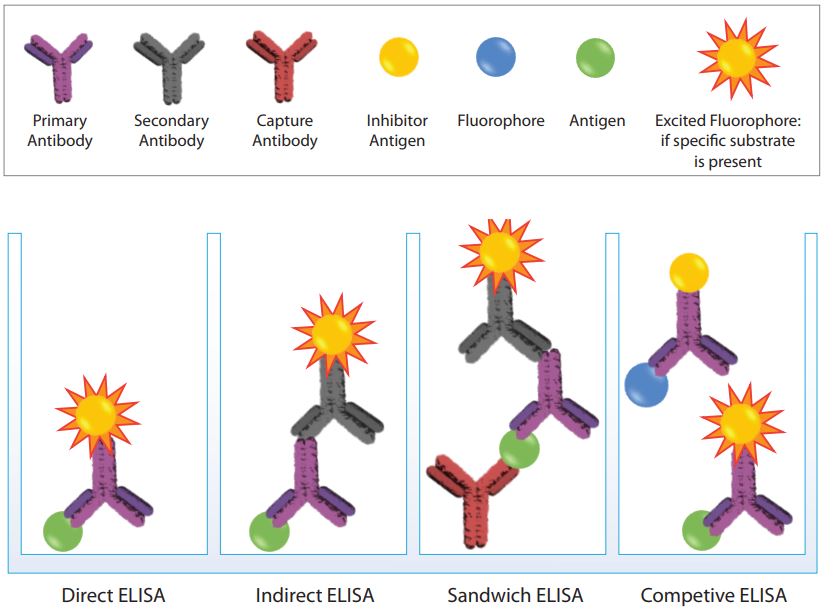
Direct ELISA
In a direct ELISA assay the antigen of interest is immobilised to the plate and detected with an enzyme-coupled antibody, while a blocking agent is used to occupy unspecific binding sites. This approach is used for the detection and quantification of non-commercial proteins.
+ Quick protocol due to the use of only one antibody.
+ No possibility of cross-reacting secondary antibody.
- Immunoreactivity of the primary antibody could be adversely affected by labelling with enzymes or tags.
- Time-consuming and expensive labelling of primary antibodies for each specific ELISA system.
- Constraint to the labelling method of the primary antibody label from experiment to experiment.
- Minimal signal amplification.
Indirect ELISA
For indirect detection, the antigen coated to a multi-well plate is detected in two stages or layers. First an unlabeled primary antibody, which is specific for the antigen, is applied. Next, an enzyme-labeled secondary antibody is bound to the first antibody. The secondary antibody is usually an anti-species antibody and is often polyclonal. The indirect assay, the most popular format for ELISA, has the following advantages and disadvantages:
+ A wide range of labeled secondary antibodies is commercially available.
+ Versatile approach, as many primary antibodies can be generated in one species and the secondary antibody, with the same label can be used for detection.
+ Highest possible immunoreactivity of the primary antibody is retained because it is not labeled.
+ Increased sensitivity as the primary antibody contains several epitopes that can be bound by the labeled secondary antibody, enhancing the signal.
- The secondary antibody might cross-react and cause a nonspecific signal.
- An additional incubation step is required for the procedure.
Sandwich ELISA
In a sandwich ELISA two antibodies, specific for different, non-overlapping epitopes are used: firstly, the plate is coated with a capture antibody. In the next step the sample is added and bound by the capture antibody. The signal identification is achieved by adding the secondary detection antibody.
+ High specificity due to the method of specifically capturing and detecting the antigen.
+ Suitable for complex (or heterogeneous/impure) samples: the antigen does not require purification prior to measurement.
+ High flexibility and sensitivity as both direct and indirect detection methods can be used.
Competitive ELISA
The method of the competitive ELISA is based on the competitive reaction between the sample antigen and the antigen bound to the microtiter plate, labeled with the primary antibody. Initially, the primary antibody is incubated with the sample antigen and the formed antibody-antigen complexes are added to the antigen coated microtiter plates. Following an incubation step, any unbound antibody is washed off.
The more antigen present in the sample, the more primary antibody will be bound to the sample antigen. Conversely, there will be a smaller amount of primary antibody available to bind to the antigen coated on the well, causing a signal reduction.
The main advantage of this ELISA method arises from its high sensitivity to compositional differences in complex antigen mixtures, even when the specific detecting antibody is present in relatively small amounts.
| |
Indirect |
Direct |
Sandwich |
Competitive |
| Capture Antibody Coating |
X |
X |
✓ |
X |
| Antigen Coating |
✓ |
✓ |
X |
✓ |
| Blocking |
✓ |
✓ |
✓ |
✓ |
| Sample (Antigen) Incubation |
X |
X |
✓ |
✓ |
| Primary Antibody Incubation |
✓ |
✓ |
✓ |
✓ |
| Secondary Antibody Incubation |
✓ |
X |
✓ |
✓ |
| Substrate Preparation |
✓ |
✓ |
✓ |
✓ |
| Signal Detection |
✓ |
✓ |
✓ |
✓ |
| Data Analysis |
✓ |
✓ |
✓ |
✓ |
4. ELISA results
The ELISA assay produces 3 types of data output:
1) Qualitative: ELISA can give some information about the presence of a particular antigen in a sample, as compared to a blank well containing no antigen or an unrelated control antigen.
2) Quantitative: the concentration of antigen in the sample is inferred from a comparison to a standard curve (serial dilution of an antigen of known concentration).
3) Semi-Quantitative: ELISAs can be used to compare the relative levels of antigen in assay samples, since the intensity of signal will vary directly with antigen concentration.
ELISA data is typically graphed with optical density against the log concentration to produce a sigmoidal curve. Known concentrations of antigen are used to generate a standard curve which is further used to measure the concentration of unknown samples by comparison to the linear portion of the standard curve.
Effectively, it is the relatively long linear region of the curve that makes the ELISA results accurate and reproducible. The unknown concentration can be determined directly on the graph or with curve fitting software, which is typically found on ELISA plate readers.

5. Sample Preparation
The procedure below provides a general guidance for the preparation of commonly tested samples for use in ELISA assays.
Generally:
- Protein extract concentration should be at 1-2 mg/mL.
- Cell and tissue extracts are diluted 1:1 with binding buffer.
- Samples are centrifuged at 10,000 rpm for 5 min at 4°C before use, to remove any precipitate.
• Cell Culture Supernatants
Centrifuge cell culture media at 1,500 rpm for 10 min at 4°C, aliquot and assay supernatant immediately. Hold at -80°C and avoiding freeze thawing.
• Cell Extracts
Place tissue culture plates on ice. Remove the media and gently wash cells once with chilled (10 mM Na2HPO4 and 1.8 mM NaH2PO4 in deionized water with 0.2% Tween 20; pH Adjusted to 7.4). Remove the PBS and add 0.5 ml extraction buffer per 100 mm plate. Tilt the plate and scrape the cells into a pre-chilled tube. Vortex briefly and incubate on ice for 15-30 min. Centrifuge at 13,000 rpm for 10 min at 4°C to pellet the insoluble content. Aliquot the supernatant in chilled tubes on ice, store at -80°C and avoid freeze thawing.
• Conditioned Media
Plate the cells in complete growth medium (with serum) until the desired level of confluence is achieved. Remove the growth media and gently wash cells twice with 2- 3 ml of warm PBS. Remove the PBS, gently add serum-free growth medium and incubate for 1-2 days. Transfer the medium into a centrifuge tube and centrifuge at 1,500 rpm for 10 min at 4°C. Aliquot the supernatant, store samples at -80°C and avoid freeze thawing.
• Tissue Extract
Homogenize tissue on ice in ice-cold buffer, containing protease inhibitors. Place the tissue in micro-centrifuge tubes and snap-freeze in liquid nitrogen. Store samples at -80°C or keep on ice for immediate homogenization. Add 300 μL of extraction buffer per 5 mg of tissue and homogenize.
• Extraction buffer
00 mM Tris, pH 7.4
150 mM NaCL
1 mM EGTA
1 mM EDTA
1% Triton X-100 0.5%
0.5% sodium deoxycholate (SDS)
Extraction buffer can be prepared in advance and stored at 4°C. Add phosphatase inhibitor (according to manufacturers guidelines), protease inhibitor (according to manufacturers guidelines) and PMSF to 1 mM before use.
Rinse the blade of the homogenizer twice with 300 μL extraction buffer. Place the sample on a shaker at 4°C for 2 hours. Centrifuge the sample for 20 min at 13,000 rpm at 4°C. Aliquot the supernatant into pre-chilled tubes on ice. Keep the samples at -80°C and avoid freeze-thawing
Note: Lysis buffer volume must be determined according to the amount of tissue present. Typical concentration of final protein extract is at least 1 mg/mL.
6. Recommended Protocols

• Reagent Preparation
Standard Solutions (use within 2 hours) For the standard concentration of 10,000 pg/mL add 1 mL of sample diluent buffer into one tube of standard (10 ng per tube) and mix thoroughly.
Note: Store this solution at 4°C for up to 12 hours (or -20°C for 48 hours) and avoid freeze-thawing.
For the standard concentration of 5,000 pg/mL add 0.3 mL of 10,000 pg/mL to 0.3 mL of sample diluent buffer and mix thoroughly.
For the standard concentration of 2,500 pg/mL add 0.3 mL of 5,000 pg/mL to 0.3 mL of sample diluent buffer and mix thoroughly.
Perform similar dilutions to obtain the standard solutions with the concentrations of 1,250, 625, 312, 156 and 78 pg/mL.
Add 100 μL of each of the diluted standard solutions to the appropriate empty wells. Repeat in duplicate or triplicate for accuracy.
Biotinylated Antibody
Calculate the total volume needed for the assay by multiplying 0.1 mL/well and the number of wells required. Add 2-3 extra wells to the calculated number of wells to account for possible pipetting errors. Generate the required volume of diluted antibody by performing a 1:100 dilution (For each 1 μL concentrated antibody, add 99 μL antibody dilution buffer) and mixing thoroughly.
Avidin-Biotin-Peroxidase (ABC)
Diluted ABC solution has to be prepared freshly. Calculate the total volume needed for the assay by multiplying 0.1 mL/well and the number of wells required. Add 2-3 extra wells to the calculated number of wells to account for possible pipetting errors. Generate the required volume of diluted ABC solution by performing a 1:100 dilution (For each 1 μL concentrated ABC solution, add 99 μL ABC dilution buffer) and mixing thoroughly.
• Direct ELISA protocol
This is a general protocol in which antigen coating and blocking may not be required if the wells have been pre-adsorbed with the antigen by the manufacturer.
Antigen Coating
- Dilute purified antigens to a final concentration of 1-10 μg/ml in bicarbonate/carbonate antigen-coating buffer (100 mM NaHCO3 in deionized water; pH adjusted to 9.6). Pipette 100 μL of diluted antigen to each well of a microtiter plate.
- Cover the plate with adhesive plastic and incubate at 4°C overnight or 37°C for 30 minutes.
- Remove the coating solution and wash the plate 3 times with 200 μL PBS buffer with for 5 minutes each time. The coating/washing solutions can be removed by flicking the plate over a sink. The remaining drops can be removed by patting the plate on a paper towel or by aspiration. Do not allow the wells to dry out at any time.
Blocking
- Pipette 200 μL blocking buffer (5% w/v non-fat dry milk in PBS buffer) per well to block residual protein-binding sites. Alternatively, BSA or BlockACE can be used to replace non-fat dry milk.
- Cover the plate with adhesive plastic and incubate for 1-2 hour(s) at 37°C (or at 4°C overnight).
- Remove the blocking solution and wash the plate twice with 200 μL PBS for 5 min each time. Flick the plate and pat the plate as described in the coating step.
Reagent Preparation
Prepare for the diluted standard solutions as shown on above.
Primary Antibody Incubation
- Serially dilute the conjugated primary antibody with blocking buffer immediately before use. The optimal dilution should be determined by a titration assay according to the antibody manufacturer.
- Pipette 100 μL of diluted secondary antibody solution to each well.
- Cover the plate with adhesive plastic and incubate for 2 hours.
- Remove the content in the wells and wash them 3 times with 200 μL PBS buffer for 5 min each time. Flick and pat the plate as described in the coating step.
Substrate Preparation
Prepare the substrate solution immediately before use or bring the pre-made substrate to room temperature. The two widely used enzymes for signal detection are horse radish peroxidase (HRP) and alkaline phosphatase (AP), and their corresponding substrates, stopping solutions, detection absorbance wavelengths and colour developed are as follows:
| Enzyme |
Substrate* |
Stop Solution |
Absorbance (nm) |
Colour Developed |
| HRP |
TMB |
2M H2SO4 |
450 |
Yellow |
| ARP |
pNPP |
0.75M NaOH |
450 |
Yellow |
| TMB: 3,3’,5,5’-tetramethylbenzidine; pNPP: p-nitrophenyl-phosphate |
Note:
• The TMB substrate must be kept at 37°C for 30 min before use.
• Hydrogen peroxide can also act as a substrate for HRP. • Sodium azide is an inhibitor of HRP. Do not include the azide in buffers or wash solutions if HRP-labeled conjugate is used for detection.
Signal Detection
- Pipette 90 μL of substrate solution to the wells with the control and standard solutions.
- Incubate the plate at 37°C in the dark. If TMB is used, shades of blue will be observed in the wells with the most concentrated solutions. Other wells may show no obvious colour.
- Colour should be developed in positive wells after 15 min. After sufficient colour development, pipette 100 μL of stopping solution to the wells (if necessary).
- Read the absorbance (OD: Optical Density) of each well with a plate reader.
Data Analysis
- Prepare a standard curve using the data produced from the diluted standard solutions. Use absorbance on the Y-axis (linear) and concentration on the X-axis (log scale).
- Interpret the sample concentration from the standard curve.
• Indirect ELISA protocol
This is a general protocol in which antigen coating and blocking may not be required if the wells from the manufacturer have been pre-adsorbed with the antigen.
Antigen Coating
- Dilute the purified antigens to a final concentration of 1-10 μg/mL in bicarbonate/carbonate antigen-coating buffer (100 mM NaHCO3 in deionized water; pH adjusted to 9.6).
- Pipette 100 μL of diluted antigen to each well of the microtiter plate.
Blocking
- Pipette 200 μL blocking buffer (5% w/v non-fat dry milk in PBS buffer) per well to block residual protein-binding sites. Alternatively, BSA or BlockACE can be used to replace the non-fat dry milk.
- Cover the plate with adhesive plastic and incubate for 1-2 hour(s) at 37°C or at 4°C overnight.
- Remove the blocking solution and wash the plate twice with 200 μL PBS for 5 minutes each time. Flick and pat the plate as described in the coating step.
Reagent Preparation
- Prepare for the diluted standard solutions as shown on above.
Primary Antibody Incubation
- Serially dilute the primary antibody of choice with blocking buffer. The optimal dilution should be determined by a titration assay according to the antibody manufacturer.
- Pipette 100 μL of each diluted antibody per well. Repeat in duplicate or triplicate for accuracy. The negative control should be species- and isotype-matched, as well as non-specific immunoglobulin diluted in PBS buffer.
- Cover the plate with adhesive plastic and incubate for 1 hour at 37°C or 2 hours at room temperature. These incubation times should be sufficient to achieve a strong signal. However, if a weak signal is observed, perform incubation overnight at 4°C to obtain a stronger signal.
- Remove the diluted antibody solution and wash the wells 3 times with 200 μL PBS for 5 minutes each time. Flick the plate and pat the plate as described in the coating step.
Secondary Antibody Incubation
- Serially dilute the conjugated secondary antibody with blocking buffer immediately before use. The optimal dilution should be determined by a titration assay according to the antibody manufacturer.
- Pipette 100 μL of diluted secondary antibody solution to each well.
- Cover the plate with adhesive plastic and incubate for 2 hours at room temperature.
- Remove the content in the wells and wash them 3 times with 200 μL PBS buffer for 5 minutes each time. Flick and pat the plate as described in the coating step.
Substrate Preparation
Prepare the substrate solution immediately before use or bring the pre-made substrate to room temperature. The two widely used enzymes for signal detection are horse radish peroxidase (HRP) and alkaline phosphatase (AP), and their corresponding substrates, stopping solutions, detection absorbance wavelengths and colour developed are as follows:
| Enzyme |
Substrate* |
Stop Solution |
Absorbance (nm) |
Colour Developed |
| HRP |
TMB |
2M H2SO4 |
450 |
Yellow |
| AP |
pNPP |
0.75M NaOH |
450 |
Yellow |
| TMB: 3,3’,5,5’-tetramethylbenzidine; pNPP: p-nitrophenyl-phosphate |
Note:
• The TMB substrate must be kept at 37°C for 30 min before use.
• Hydrogen peroxide can also act as a substrate for HRP.
• Sodium azide is an inhibitor of HRP. Do not include the azide in buffers or wash solutions if HRP-labeled conjugate is used for detection.
Sandwich ELISA protocol
Capture Antibody Coating
- Dilute the capture antibody to a final concentration of 1-10 μg/mL in bicarbonate/carbonate antigen-coating buffer (100 mM NaHCO3 in deionized water; pH adjusted to 9.6).
- Pipette 100 μL of diluted antibody to each well of a microtiter plate.
- Cover the plate with adhesive plastic and incubate at 4°C overnight (or 37°C for 30 min).
- Remove the coating solution and wash the plate 3 times with 200 μL PBS with for 5 minutes each time. The coating/washing solutions can be removed by flicking the plate over a sink. The remaining drops can be removed by patting the plate on a paper towel or by aspiration. Do not allow the wells to dry out at any time.
Blocking
- Pipette 200 μL blocking buffer (5% w/v non-fat dry milk in PBS buffer; Alternatively, BSA or BlockACE can be used to replace non-fat dry milk.) per well to block residual protein-binding sites.
- Cover the plate with adhesive plastic and incubate for 1-2 hour(s) at 37°C (or at 4°C overnight).
- Remove the blocking solution and wash the plate twice with 200 μL PBS for 5 minutes each time. Flick and tap the plate as described in the coating step.
Reagent Preparation
Prepare biotinylated antibody and ABC solutions as described previously for the diluted standard solutions.
Sample (Antigen) Incubation
- Serially dilute the sample with blocking buffer immediately before use. The optimal dilution should be determined by a titration assay according to the antibody manufacturer.
- Pipette 100 μL of each of the diluted sample solutions and control to each empty well. Repeat in duplicate or triplicate for accuracy. The negative control should be species- and isotype-matched, as well as non-specific immunoglobulin diluted in PBS buffer.
- Cover the plate with adhesive plastic and incubate for 2 hours at room temperature.
- Remove the content in the wells and wash them 3 times with 200 μL PBS buffer for 5 minutes each. Flick the plate and pat the plate as described in the coating step.
Biotinylated Antibody Incubation
- Pipette 100 μL of diluted antibody to the control, standard solutions and diluted samples.
- Cover the plate with adhesive plastic and incubate for 1 hour at 37°C (or 2 hours at room temperature). These incubation times should be sufficient to receive a strong signal. However, if a weak signal is observed, perform incubation overnight at 4°C for a stronger signal.
- Remove the content in the wells and wash them 3 times with 200 μL PBS for 5 min each time. Flick the plate and pat the plate as described in the coating step.
ABC Incubation
- Pipette 100 μL of diluted ABC solution to the wells with control, standard solutions and diluted samples.
- Cover the plate with adhesive plastic and incubate for 30 minutes at 37°C.
- Remove the content in the wells and wash them 3 times with 200 μL PBS buffer for 5 minutes each. Flick the plate and pat the plate as described in the coating step.
Substrate Preparation
Prepare the substrate solution immediately before use or bring the pre-made substrate to room temperature. The two widely used enzymes for signal detection are horse radish peroxidase (HRP) and alkaline phosphatase (AP), and their corresponding substrates, stopping solutions, detection absorbance wavelengths and colour developed are as follows:
| Enzyme |
Substrate* |
Stop Solution |
Absorbance (nm) |
Colour Developed |
| HRP |
TMB |
2M H2SO4 |
450 |
Yellow |
| AP |
pNPP |
0.75M NaOH |
450 |
Yellow |
| TMB: 3,3’,5,5’-tetramethylbenzidine; pNPP: p-nitrophenyl-phosphate |
Note:
• The TMB substrate must be kept at 37°C for 30 minutes before use.
• Hydrogen peroxide can also act as a substrate for HRP.
• Sodium azide is an inhibitor of HRP. Do not include the azide in buffers or wash solutions if HRP-labeled conjugate is used for detection.
Signal Detection
- Pipette 90 μL of substrate solution to the wells with the control, standard solutions and diluted samples.
- Incubate the plate at 37°C in the dark. If TMB is used, shades of blue will be observed in the wells with the most concentrated solutions. Other wells may show no obvious colour.
- A colourimetric signal should be developed in positive wells after 15 minutes. Stop the reaction by adding 100 μL of stop solution.
- Read the absorbance (OD: Optical Density) of each well with a plate reader.
Data Analysis
- Prepare a standard curve using the data produced from the diluted standard solutions. Use absorbance on the Y-axis (linear) and concentration on the X-axis (log scale). Competitive ELISA yields an inverse curve: Higher values of antigen in the samples yield a smaller amount of colour change.
- Interpret the sample concentration from the standard curve.
• Competitive ELISA protocol
This is a general protocol in which antigen coating and blocking may not be required if the wells have been pre-adsorbed with the antigen by the manufacturer.
Antigen Coating
- Dilute purified antigens to a final concentration of 20 μg/ml in bicarbonate/carbonate antigen-coating buffer (100 mM NaHCO3 in deionized water; pH adjusted to 9.6).
- Pipette 100 μL of diluted antigen to each well of a microtiter plate.
- Cover the plate with adhesive plastic and incubate at 4°C overnight or 37°C for 30 minutes.
- Remove the coating solution and wash the plate 3X with 200 μL PBS buffer with for 5 minutes each time. The coating/washing solutions can be removed by flicking the plate over a sink. The remaining drops can be removed by patting the plate on a paper towel or by aspiration. Do not allow the wells to dry out at any time.
Blocking
- Pipette 200 μL blocking buffer (5% w/v non-fat dry milk in PBS buffer) per well to block residual protein-binding sites. Alternatively, BSA or BlockACE can be used to replace non-fat dry milk.
- Cover the plate with adhesive plastic and incubate for 1-2 hour(s) at 37°C or at 4°C overnight.
- Remove the blocking solution and wash the plate twice with 200 μL PBS for 5 minutes each time. Flick and pat the plate as described in the coating step.
Reagent Preparation - Prepare for the diluted standard solutions as shown on p.9.
Sample Incubation
- Serially dilute the sample with blocking buffer immediately before use. The optimal dilution should be determined by a titration assay according to the antibody manufacturer.
- Pipette 100 μL of diluted sample to each well.
- Cover the plate with adhesive plastic and incubate for 2 hours at room temperature.
- Remove the content in the wells and wash them three times with 200 μL PBS buffer for 5 minutes each time. Flick and pat the plate as described in the coating step.
Primary Antibody Incubation
- Serially dilute the primary antibody of choice with blocking buffer. The optimal dilution should be determined by a titration assay according to the antibody manufacturer.
- Pipette 100 μL of each diluted antibody per well. Repeat in duplicate or triplicate for accuracy. The negative control should be species- and isotype-matched as well as non-specific immunoglobulin diluted in PBS buffer.
- Cover the plate with adhesive plastic and incubate for 1 hour at 37°C or 2 hours at room temperature. These incubation times should be sufficient to receive a strong signal. However, if a weak signal is observed, perform incubation overnight at 4°C for a stronger signal.
- Remove the diluted antibody solution and wash the wells 3 times with 200 μL PBS for 5 minutes each time. Flick the plate and pat the plate as described in the coating step.
Secondary Antibody Incubation
- Serially dilute the conjugated secondary antibody with blocking buffer immediately before use. The optimal dilution should be determined by a titration assay according to the antibody manufacturer.
- Pipette 100 μL of diluted secondary antibody solution to each well.
- Cover the plate with adhesive plastic and incubate for 2 hours at room temperature.
- Remove the content in the wells and wash them 3 times with 200 μL PBS buffer for 5 minutes each time. Flick the plate and pat the plate as described in the coating step.
Substrate Preparation
Prepare the substrate solution immediately before use or bring the pre-made substrate to room temperature. The two widely used enzymes for signal detection are horse radish peroxidase (HRP) and alkaline phosphatase (AP), and their corresponding substrates, stopping solutions, detection absorbance wavelengths and colour developed are as follows:
| Enzyme |
Substrate* |
Stop Solution |
Absorbance (nm) |
Colour Developed |
| HRP |
TMB |
2M H2SO4 |
450 |
Yellow |
| AP |
pNPP |
0.75M NaOH |
450 |
Yellow |
| TMB: 3,3’,5,5’-tetramethylbenzidine; pNPP: p-nitrophenyl-phosphate |
Note:
• The TMB substrate must be kept at 37°C for 30 minutes before use.
• Hydrogen peroxide can also act as a substrate for HRP.
• Sodium azide is an inhibitor of HRP. Do not include the azide in buffers or wash solutions if HRP-labeled conjugate is used for detection.
Signal Detection
- Pipette 90 μL of substrate solution to the wells with the control, standard solutions and diluted samples.
- Incubate the plate at 37°C in the dark. If TMB is used, shades of blue will be observed in the wells with the most concentrated solutions. Other wells may show no obvious colour.
- Colour should be developed in positive wells after 15 minutes. After sufficient colour development, pipette 100 μL of stopping solution to the wells (if necessary).
- Read the absorbance (OD: Optical Density) of each well with a plate reader.
Data Analysis
- Prepare a standard curve using the data produced from the diluted standard solutions. Use absorbance on the Y-axis (linear) and concentration on the X-axis (log scale).
- Competitive ELISA yields an inverse curve: Higher values of antigen in the samples yield a smaller amount of colour change.
- Interpret the sample concentration from the standard curve
7. Troubleshooting Guide
The following guide serves as a checklist for the possible causes and solutions with respect to some of the most commonly encountered problems from the ELISA assays.
• Weak / no signal
| |
Possible cause |
Solution to the problem |
| 1 |
Blocking protein in coating solution |
Eliminate blocking protein from coating solution |
| 2 |
Capture antibody (or antigen) does not bind to plate |
Use ELISA plate, not tissue culture plate Extend coating time Increase concentration of coating components |
| 3 |
Problem with the standard |
Use new sample Check that the standard is appropriately handled |
| 4 |
Shortened incubation time |
Follow the manufacturer guideline (If the problem persists, try incubating samples at 4°C overnight) |
| 5 |
Incubation temperature too low |
Ensure incubations are done at correct temperature Before proceeding, all reagents, including plate, should be at room temperature or as recommended by the manufacturer |
| 6 |
Incompatible sample type |
Use sample that the assay is known to detect a positive control (Include such control in your experiment) |
| 7 |
Incompatible assay buffer |
Ensure assay buffer is compatible with the target of interest |
| 8 |
Target present below detection limit |
Decrease dilution factor or concentrate samples |
| 9 |
Incorrect/Insufficient/No substrate |
Check the substrate identity Increase concentration or amount of substrate Follow manufacturer guidelines |
| 10 |
Incorrect/Insufficient/No antibody |
Check the antibody identity Repeat the assay with higher antibody concentrations to find the optimal one for your experiment |
| 11 |
Antibody stored at 4°C for several weeks or subjected to repeated freeze-thaw cycles |
Use fresh aliquot of antibody that has been stored at -20°C or below |
| 12 |
Incorrect reagents added/ prepared; Missing reagents |
Check protocol, ensure correct reagents are added in proper order and prepared to correct concentrations (e.g. TMB for HRP-labeled antibodies) |
| 13 |
Expired/Contaminated reagents |
Make and use fresh/uncontaminated reagents |
| 14 |
Enzyme inhibitor present |
Avoid sodium azide in HRP reactions Avoid phosphate in AP reactions |
| 15 |
Incorrect storage of components |
Double check storage conditions on kit level (Most kits need to be stored at 4°C) |
| 16 |
Ultra-vigorous plate washing |
Gently pipette wash buffer (manual method) Ensure correct pressure (automatic wash system) |
| 17 |
Dried out wells |
Cover the plate using sealing film or tape for all incubations |
| 18 |
Wells scratched with pipette or pipette tips |
Carefully dispense/aspirate solutions into and out of wells |
| 19 |
Plate read at incorrect detection wavelength |
Use recommended wavelength/filter Ensure plate reader is set correctly for type of substrate used |
| 20 |
Slow colour development |
Prepare substrate immediately before use Allow longer incubation Ensure stock solution is unexpired and uncontaminated |
| 21 |
Epitope recognition impeded by adsorption to plate |
Conjugate peptide to large carrier protein before coating onto plate |
• Saturated signal
| |
Possible cause |
Solution to the problem |
| 1 |
High sample concentration |
Use higher sample dilutions (Determine the optimal dilutions by titration assay) |
| 2 |
Excessive substrate |
Decrease concentration or amount of substrate: Follow manufacturer guidelines (The substrate provided with the ELISA kit might require further dilution) |
| 3 |
Substrate colour changed before |
Make substrate immediately before use |
| 4 |
Unspecific antibody binding |
Ensure wells are pre-processed to prevent non-specific binding.
Use affinity-purified antibody and preferably one that is pre-adsorbed.
Use serum (5-10%) from same species as secondary antibody (bovine serum is also recommended) |
| 5 |
Exceeded incubation time |
Follow the manufacturer guidelines (If the problem persists, try incubating samples at 4°C overnight) |
| 6 |
Excess antibody |
Repeat the assay with lower antibody concentrations to find the optimal one for your experiment |
| 7 |
Contaminated buffers with metals or HRP |
Make and use fresh buffers |
| 8 |
Insufficient washing |
Follow the manufacturer guidelines At the end of each washing step, flick the plate over a sink and pat the plate on a paper towel |
| 9 |
Plate sealers not used or re-used |
Use recommended wavelength/filter.
Ensure plate reader is set correctly for type of substrate used. |
| 10 |
Plate read at incorrect detection wavelength |
Conjugate peptide to large carrier protein before coating onto plate |
| 11 |
Excess time before plate reading |
Read your plate within 30 minutes after adding the substrate (If the reading is not performed within this time frame, add a stopping solution after sufficient colour is developed in the plate) |
• High Background
| |
Possible cause |
Solution to the problem |
| 1 |
Insufficient washing |
Follow the manufacturer guidelines
At the end of each washing step, flick the plate over a sink and pat the plate on a paper towel |
| 2 |
Ineffective/Contaminated blocking buffer |
Try higher blocking protein concentration
Increase blocking time
Use fresh buffer |
| 3 |
Excess antibody |
Repeat the assay with lower antibody concentrations to find the optimal one for your experiment |
| 4 |
Excess substrate |
Decrease concentration or amount of substrate Follow manufacturer guidelines (Note: The substrate provided with the ELISA kit might require further dilution) |
| 5 |
Cross reactivity (Detection antibody reacts with coating antibody) |
Run appropriate controls |
| 6 |
Unspecific antibody binding |
Try different formulations in coating solutions
Ensure wells are pre-processed to prevent non-specific binding
Use affinity-purified antibody and preferably one that is pre-adsorbed
Use serum (5-10%) from same species as secondary antibody (bovine serum is also recommended) |
| 7 |
Insufficient Tween in buffers |
Use PBS containing 0.05% Tween |
| 8 |
Suboptimal salt concentration in washing buffer |
Optimize salt concentration as high concentration can reduce non-specific interactions |
| 9 |
Exceeded incubation temperature |
Optimize incubation temperature for your assay (antibodies bind optimally at very specific temperature) |
| 10 |
Insufficient reagent mixture |
Thoroughly mix all reagents and samples before pipetting solutions into wells |
| 11 |
Blanks contaminated with samples |
Change pipette tips when switching between blanks and samples
Put a lid on plates to avoid any spilling between wells |
| 12 |
Sample contaminated with enzymes |
Test samples with substrate alone to check for contaminating enzymes |
| 13 |
Contaminated TMB substrate |
Use a clean container to check that the substrate is not contaminated (TMB substrate should be clear and colourless before adding to wells) |
| 14 |
Substrate incubation in light |
Carry out substrate incubation in dark or follow recommendation from manufacturer |
| 15 |
Uneven evaporation of solution from wells during incubation |
Always incubate with a lid on the plate |
| 16 |
Precipitate created in wells upon substrate addition |
Increase dilution factor of sample or decrease concentration of substrate |
| 17 |
Exceeded incubation time |
Follow the manufacturer guidelines (If the problem persists, try incubating samples at 4°C overnight) |
| 18 |
Incorrect standard curve dilutions |
Check pipetting techniques.
Double check calculations. |
| 19 |
Plates stacked during incubations, leading to uneven temperature distribution |
Avoid stacking plates |
| 20 |
Dirty or defective plates |
Clean the plate bottom |
| 21 |
Unstopped colour development |
Use Stopping solution to prevent over-development |
| 22 |
Excess time before plate reading |
Read your plate within 30 minutes after adding the substrate (If the reading is not performed within this time frame, add a stopping solution after sufficient colour is developed in the plate)
Note: Colour continues to develop even after adding the stopping solution (although at a slower rate) |
| 23 |
Incorrect plate reading setting |
Use recommended wavelength/filter
Ensure plate reader is set correctly for type of substrate used |
• Low sensitivity
| |
Possible cause |
Solution to the problem |
| 1 |
Assay format not sensitive enough |
Switch to a more sensitive detection system (e.g. colourimetric to chemiluminescence)
Switch to a more sensitive assay type (e.g. direct ELISA to sandwich ELISA)
Increase incubation time and/or temperature |
| 2 |
Improper storage of ELISA kit |
Store all reagents as recommended (Note: all reagents may not have identical storage requirements) |
| 3 |
Insufficient target |
Reduce sample dilution or concentrate sample |
| 4 |
Inactive substrate |
Ensure reporter enzyme has the expected activity |
| 5 |
Poor target adsorption to wells |
Covalently link target to wells |
| 6 |
Insufficient substrate |
Increase concentration or amount of substrate |
| 7 |
Incompatible sample type |
Use a sample that the assay is known to detect a positive control.
Include positive control in your experiment. |
| 8 |
Interfering ingredients in buffers and sample |
Check reagents for any interfering chemicals, e.g. sodium azide in antibodies inhibit HRP enzyme; EDTA used as anti-coagulant for plasma collection inhibits enzymatic reactions |
| 9 |
Mixing or substituting reagents from different kits |
Avoid mixing components from different kits |
| 10 |
Incorrect plate reading setting |
Use recommended wavelength/filter Ensure plate reader is set correctly for type of substrate used |
• Poor standard curve
| |
Possible cause |
Solution to the problem |
| 1 |
Improper standard solution |
Confirm dilutions are done correctly.
Make new standard curve as appropriate. |
| 2 |
Standard improperly reconstituted |
Briefly spin vial before opening.
Inspect for undissolved material after reconstituting. |
| 3 |
Standard degraded |
Store and handle standard as recommended.
Prepare standards no more than two hours before use. |
| 4 |
Improper curve fitting |
Try plotting using different scales, e.g. log-log, 5-parameter logistic curve fit |
| 5 |
Pipetting error |
Use calibrated pipettes and proper pipetting technique |
| 6 |
Insufficient washing |
Follow the manufacturer guidelines. At the end of each washing step, flick the plate over a sink and pat the plate on a paper towel |
| 7 |
Insufficiently mixed reagents |
Thoroughly mix reagents |
| 8 |
Poor/variable adsorption of reagents to plate |
Extend incubation time.
Check coating buffer.
Use a different plate as appropriate.
Check homogeneity of samples. |
| 9 |
Plates stacked during incubation |
Keep plates separated if not using rotating plates |
| 10 |
Dirty or defective plates |
Clean the plate bottom |
• Poor replicate data
| |
Possible cause |
Solution to the problem |
| 1 |
Bubble in wells |
Ensure no bubbles are present prior to reading plate |
| 2 |
Insufficient washing of wells |
Carefully wash wells.
Follow recommended protocols.
Check that all ports of the plate washer are unobstructed. |
| 3 |
Incomplete reagent mixing |
Ensure all reagents are mixed thoroughly |
| 4 |
Inconsistent pipetting |
Use calibrated pipettes and proper pipetting techniques.
If a multi-channel pipette is used, ensure that all channels deliver the same volume. |
| 5 |
Inconsistent sample prep or storage |
Ensure consistent sample prep and optimal sample storage conditions (e.g. minimize freeze/thaw cycles) |
| 6 |
Particulates in samples |
Remove the particulates by centrifugation |
| 7 |
Plate sealers not used or re-used |
During incubations, cover plates with plate sealers.
Use a fresh sealer every time the used sealer is removed from the plate. |
| 8 |
Cross-well contamination |
Ensure plate sealers and pipette tips are not contaminated with reagents |
| 9 |
Edge effect (higher or lower OD in peripheral wells than in central wells) |
Ensure plates and reagents are kept at room temperature before pipetting into wells unless otherwise instructed.
During incubation, seal the plate completely with a plate sealer and avoid stacking plates. |
• Inconsistent Assay-to-Assay results
| |
Possible cause |
Solution to the problem |
| 1 |
Insufficient washing of wells |
Carefully wash wells.
Follow recommended protocols.
Check that all ports of the plate washer are unobstructed. |
| 2 |
Variation in incubation temperature |
Adhere to recommended incubation temperature.
Avoid incubating plates in area where environmental conditions vary. |
| 3 |
Variation in protocol |
Adhere to the same protocol from run to run |
| 4 |
Plate sealers not used or re-used |
During incubations, cover plates with plate sealers.
Use a fresh sealer every time the used sealer is removed from the plate. |
| 5 |
Incorrect dilutions |
Confirm dilutions are done correctly for standard solutions, etc.
Make new standard curve as appropriate. |
| 6 |
Contaminated buffers |
Make and use fresh buffers |
| 7 |
Plates stacked during incubation |
Keep plates separated if not using rotating plates |
• Slow colour development
| |
Possible cause |
Solution to the problem |
| 1 |
Substrates too old, contaminated or used at incorrect pH |
Make and use fresh substrates at correct pH: they should be prepared immediately before use |
| 2 |
Expired/Contaminated solutions |
Make and use fresh reagents |
| 3 |
Incorrect incubation temperature |
Ensure plates and reagents are kept at room temperature before pipetting into wells unless otherwise instructed.
During incubation, seal the plate completely with a plate sealer and avoid stacking plates. |
| 4 |
Low antibody concentration |
Repeat the assay with higher antibody concentrations to find the optimal one for your experiment |
| 5 |
Low substrate concentration |
Add more substrate to the wells.
Make substrate no more than one hour before use.
Note: Typical ELISA sensitivity is ~0.1 pg/mL with exact value depends on antibody used. |
• Plate imaging problems
| |
Possible cause |
Solution to the problem |
| 1 |
Oversaturated image after acquisition |
Use full resolution image to analyze results (Do not use jpeg or other compressed formats) |
| 2 |
Blurry spots in images |
Re-focus your camera before taking a new image |
| 3 |
Repeated pixel values or rectangular spots |
Use lower bin size, higher image resolution and/or lossless file type sealer and avoid stacking plates |
| 4 |
Flat standard in images |
Reduce acquisition time |
Do you have any issues with your ELISA? – contact us at Info@2BScientific.com
Gain Data
Arigo is proud to launch an in-house developed GainDataTM (ELISA analysis tool, web based free service).
Researchers just simply copy their ELISA data from excel and paste to our GainDataTM then follow the tool, less than a minute the result will be calculated automatically.